
Aspects génétiques de la mucoviscidose
La mucoviscidose (ou fibrose kystique du pancréas) est une maladie génétique héréditaire à transmission autosomique récessive. C’est à dire que seuls les sujets ayant hérité de deux mutations – l’une provenant du père, l’autre de la mère – sont atteints.
Le gène responsable de la maladie, appelé gène CFTR (Cystic Fibrosis Transmembrane Conductance Regulator) a été identifié en 1989. Il est situé sur le bras long du chromosome 7 (7q31) et code pour la protéine CFTR. Cette dernière intervient dans la régulation du transport des ions chlorures au niveau de la membrane cellulaire. Jusqu’à ce jour, plus de 2 000 mutations ont été identifiées, parmi lesquelles la plus fréquente est la mutation F508del.On la retrouve en effet chez environ 80 % des malades en France.
Dépistage de la mucoviscidose
Symptômes et test de la sueur
Avant la mise en place du dépistage néonatal systématique, le diagnostic était précédé d’une période d’errance diagnostique plus ou moins longue.
Il était finalement le plus souvent évoqué devant des signes d’appels cliniques (ileus méconial, diarrhée graisseuse, encombrement et/ou infections récidivantes des voies respiratoires) puis confirmé par un test de la sueur positif. Ce test de la sueur révélait alors un taux élevé d’ions chlorure dans la sueur. On complétait enfin les investigations par l’analyse moléculaire du gène CFTR et la recherche des mutations en cause.
Un dépistage néonatal systématique
Depuis 2002, le dépistage néonatal systématique a été étendu à l’ensemble du territoire métropolitain ainsi qu’en France d’Outre-mer. Le ministère de la Santé en a confié la prise en charge à l’Association Française pour le Dépistage et la Prévention des Handicaps de l’Enfant (AFDPHE). L’algorithme du dépistage fait appel au dosage sanguin de la trypsine immuno-réactive (TIR) et à la recherche des mutations CFTR les plus fréquentes (30 puis 29 depuis le 01/01/2015). La TIR est une protéine dont la présence est plus abondante en cas d’anomalie pancréatique pendant la vie fœtale et les premiers mois de vie. Son dosage permet donc de repérer environ 95% des nouveau-nés atteints de mucoviscidose. Toutefois, la spécificité insuffisante du dosage de la TIR (qui sélectionne également des enfants qui ne sont pas atteints de mucoviscidose) explique la nécessité du couplage à l’analyse moléculaire.
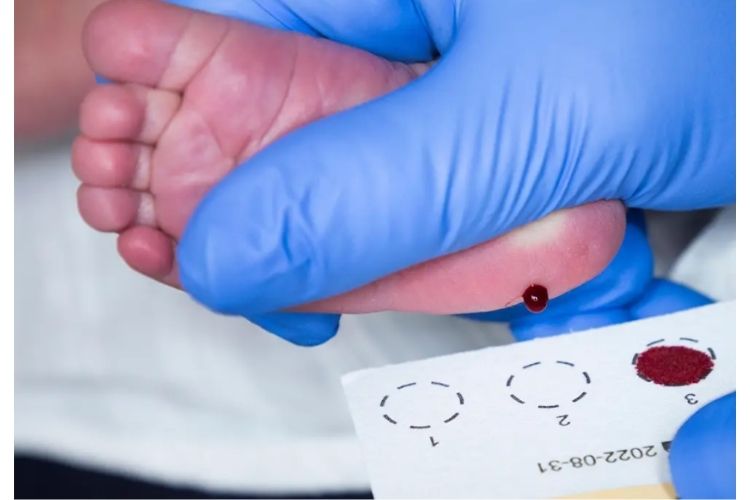
Après recherche des principales mutations CFTR, trois cas de figure peuvent se présenter :
Deux mutations sont identifiées
Le nouveau-né et ses parents sont convoqués dans un centre de ressources et de compétences de la mucoviscidose (CRCM) pour une confirmation du diagnostic. Ce diagnostic repose sur l’évaluation clinique et un test de la sueur positif, ainsi que pour la mise en place du traitement et du suivi.
Une seule mutation est identifiée
(le risque qu’une deuxième mutation ne soit pas identifiée est d’environ 10%)
Le test de la sueur doit être réalisé dans un centre spécialisé. Si le test est positif, l’enfant est pris en charge comme ceux du groupe précédent. Si le test se révèle négatif, l’information sur l’hétérozygotie du nouveau-né sera donnée aux parents lors d’une consultation de conseil génétique.
Aucune mutation n’est retrouvée et la TIR est très élevée
Un contrôle de TIR par prélèvement sanguin sur buvard vers 21 jours de vie est pratiqué. La persistance d’une TIR élevée à J21 conduira à une consultation dans un centre spécialisé pour évaluation complémentaire (test de la sueur). Un test de la sueur dont le résultat est douteux (« intermédiaire ») devra alors être répété.
Manifestations de la maladie et traitements dans la mucoviscidose
La mucoviscidose s’exprime de diverses façons
L’anomalie de fonctionnement de CFTR s’exprime principalement au niveau des voies respiratoires, du tube digestif, du foie, des glandes sudoripares et du tractus génital. Néanmoins, d’un patient à l’autre, on observe une grande diversité d’expression clinique, tant pour l’âge d’apparition des premiers symptômes que pour la sévérité de l’évolution. La sévérité de l’atteinte respiratoire conditionne le pronostic vital dans la majorité des cas.
Traiter les symptômes… et un jour les causes de la mucoviscidose
Les traitements symptomatiques – très contraignants – reposent tout d’abord sur la prise en charge respiratoire (kinésithérapie, traitements inhalés, antibiothérapie, oxygénothérapie). Ils concernent aussi la prise en charge digestive et nutritionnelle (extraits pancréatiques et régime alimentaire). La transplantation pulmonaire est le traitement de dernier recours en situation d’insuffisance respiratoire grave. Depuis quelques années, les « nouvelles thérapies » ciblées sur les dysfonctions liées à certaines mutations de CFTR (traitements modulateurs ou correcteurs) cherchent à corriger la cause de la maladie.
Il est à noter que l’éducation thérapeutique fait partie intégrante de la prise en charge multidisciplinaire.